
Cymbalta (duloxetine) : études d'efficacité
Le Cymbalta - chlorhydrate de duloxétine - est, depuis 2004, commercialisé par le laboratoire Eli Lilly principalement comme antidépresseur. Il est le second antidépresseur de type ISRNa (61) après l'Effexor - venlafaxine - de Pfizer commercialisé depuis 1994. Le Cymbalta a été synthétisé à la fin des années 1990 par le laboratoire Eli Lilly. Lilly détenait depuis 1986 le brevet du Prozac-fluoxétine, succès commercial majeur pour le premier des produits de type ISRS (61). La fin de validité du brevet du Prozac était prévue en 2001. Lilly dépose une demande d'autorisation à la Food and Drugs Administration (FDA) pour le Cymbalta en 2001, parallèlement à une demande d'extension de la durée du brevet du Prozac. Nous faisons dans cet article la synthèse de toutes les informations disponibles sur ce produit, en termes d'efficacité et d'effets secondaires/indésirables.
I. Histoire et efficacité de la duloxétine sur le trouble dépressif majeur
Nous démontrons dans cet article que Lilly n'est jamais parvenu à montrer clairement une supériorité du Cymbalta (duloxétine) sur le placebo. Les différents moyens utilisés pour masquer la pauvreté des résultats sont mis en évidence.
Le dossier initial
- 2 études (HMBHa et HMBHb) comparant la diminution du score de l'échelle de dépression Hamilton à 17 points (HAMD-17) de 60mg par rapport au placebo, 9 semaines après l'introduction,
- 1 étude (HMATa) comparant la diminution du score de l'échelle de dépression Hamilton à 17 points (HAMD-17) de 2x20mg de par rapport au placebo et à 20 mg de paroxetine (Deroxat@), 8 semaines après l'introduction,
- 1 étude (HMATb) comparant la diminution du score de l'échelle de dépression Hamilton à 17 points (HAMD-17) de 2x20mg ou 2x40mg par rapport au placebo et à 20 mg de paroxetine, 8 semaines après l'introduction.
Il a été déposé à la FDA fin novembre 2001 pour l'indication de trouble dépressif majeur, accompagné des résultats des essais suivants, menés aux USA par le laboratoire :
Au total ces études randomisées et en double aveugle portaient sur 1219 personnes vivant aux USA.
Les critères d'exclusion au départ de l'étude
- les personnes ayant des comorbidités addictives, ou d'autres troubles psychiatriques que le trouble dépressif majeur, y compris les troubles de la personnalité, un trouble anxieux, la dysthymie, etc., ou des maladies somatiques.
- les personnes pour lesquelles deux antidépresseurs n'avaient pas fonctionné précédemment, ou ayant subi des séances d'electroconvulsivotherapie (électrochocs),
- les personnes ayant un "risque de comportement suicidaire",
- d'autres exclusions, par exemple le fait d'être hospitalisé : ces exclusions sont probables et apparaîtront dans l'analyse des études ultérieures demandées, comme les études complémentaires HMAYa et HMAYb, demandées ultérieurement par la FDA, et relatées ci-après (56).
Ils ont été décrits de manière complète en 2005 dans une documentation de l'agence européenne (60), page 20. Sont ainsi exclues par exemple :
Les critères d'exclusion en cours d'étude
De plus, les résultats ne prenaient pas en compte les personnes ayant cessé de participer à l'étude avant les 8 ou 9 semaines : 30% pour le groupe "duloxétine", essentiellement pour cause de manque d'efficacité ou d'effet secondaire (60), davantage lorsque l'on se réfère aux informations échangées entre le laboratoire et la FDA (Tableau 3), avec une proportion moindre pour le placebo. Des recherches ultérieures ordonnées par différentes instances gouvernementales permettront de produire à partir de 2003 des résultats plus précis, avec ou sans prise en compte des abandons, mais ne figurent pas, pour la plupart, dans les résultats et dans les fréquences d'effets secondaires publiés jusqu'à aujourd'hui dans les notices officielles (2)(6)(46).
Revenir au début
Les résultats de 2001
Ces premiers résultats donnaient pour 3 études sur 4 un avantage entre 1 et 4 points à la duloxétine par rapport au placebo (Tableau 1) ; le résultat précis de la troisième étude (HMATa) n'est pas publié, il est simplement indiqué "l'absence d'avantage" par rapport au placebo. Dans son rapport à la FDA, Lilly indique que l’étude HMATa « a échoué ». Les chiffres des analystes de la FDA montrent que cette étude montre en réalité un légère supériorité de la paroxétine sur la duloxétine (56). La différence par rapport à la paroxétine comparée dans les études HMATa et HMATb, est généralement mineure et n’est publiée dans aucune notice officielle.

Tableau 1 : les résultats fournis par le laboratoire Lilly en 2001 à la FDA, sauf ceux de l'étude HMATa, en terme de diminution comparée du score de dépression HAMD-17 entre duloxetine et placebo à 2 mois, exclusion faite des abandons en cours d'étude - 35% à 42%. Le laboratoire estime la marge d'erreur à 0,7 points. Source : (6).
Discussion
- Une différence d'un tiers de l'écart type entre duloxétine et placebo est généralement considérée comme probante ; les chiffres ci-dessus sont donc indicateurs d'une certaine efficacité pour 2 études sur 4.
- Le placebo est très efficace dans les troubles dépressifs majeurs. Il serait intéressant de disposer d'études "placebo vs aucun médicament". Avoir une efficacité sensiblement supérieure permet d'accélérer un processus humain de rémission, qui est d'abord fondé sur le soin reçu et perçu comme reçu.
- Le très grand nombre d'abandons en cours d'étude - 35% à 42% pour les personnes sous duloxetine - laisse supposer que la prise en compte de ces abandons par une méthode de correction statistique de type LOCF aurait considérablement diminué l'efficacité statistique du Cymbalta, et aurait augmenté les incidences d'effets secondaires observés. L'agence européenne EMA indique à ce sujet que "sachant que la plupart des patients interrompent la thérapie du fait des effets secondaires, et que le manque d'efficacité est le motif habituel d'arrêt des personnes sous placebo, la correction LOCF n'aurait pas été une approche fidèle". Il est permis de penser qu'au contraire, inclure 30% de réponses supplémentaires aurait permis de mesurer en quoi l'effet secondaire ou le manque d'efficacité diminue le résultat réel des personnes sélectionnées pour l'étude. Les exclure revient à mesurer un effet artificiellement positif pour la duloxetine ou le placebo, avec une erreur de 30% pour la première. Ceci sera confirmé par la suite (Tableau 2).
- L'exclusion de l'étude HMATa de toute publication chiffrée fausse les méta-analyses qu'effectueront par la suite les chercheurs.
- Certains chercheurs pensent que l'aspect "double-aveugle" est fictif, dans la mesure où la présence de nombreux effets secondaires rend visible le fait de prendre la molécule, et donc peut favoriser une notation subjective positive de son efficacité (biais de mesure). D'autres rappellent que dans l'autre sens, l'effet "nocebo" tend à une déclaration d'effets secondaires amplifiée pour des raisons psychologiques, et de moindre efficacité. Toutefois l'effet "nocebo" joue autant pour la molécule que pour le placebo.
- Fait notable, l’analyse détaillée de l’étude HMHBb a montré une légère supériorité du placebo pour les personnes se plaignant d’insomnies, ainsi que pour les cas de dépression mélancolique, tandis que les autres études ne relevaient pas ce point (56).
- Enfin, tandis que les mesures réalisées à l’aide de l’échelle HAMD17 et de l’échelle CGI (Clinical Global Improvment, basée sur un questionnaire rempli par le praticien) montraient une supériorité relative (hors abandons) pour 2 études sur 4, l’analyse des résultats sur l’échelle PGI (Patient Global Improvment, basée sur un questionnaire rempli par le patient), montraient au contraire une supériorité du placebo : légère pour les doses de 40mg, probante pour les doses de 60mg (Tableau 3, ci-après). (56). Ces résultats sur l’échelle PGI n’ont pourtant donné lieu à aucun commentaire, et ne figurent pas dans les notices officielles finales.
Revenir au début
Les résultats complémentaires de 2003
La FDA, au vu de la demande initiale du laboratoire et des données présentées, répondit le 13 septembre 2002 par un avis provisoire positif, assorti d’une demande de précisions préalables à un avis d’autorisation. La FDA relevait que les doses de plus de 60mg ne montraient pas une efficacité supérieure, et demandait donc des précisions sur ce point, et une recommandation étayée pour la dose de départ (56).
Le laboratoire contesta que les doses de plus de 60mg n’apportaient pas un bénéfice supplémentaire, et mena deux nouvelles études en Europe de l’Est (Bulgarie, Roumanie, Russie, Hongrie, Pologne et Slovaquie), les études HMAYa et HMAYb, avec cette fois des doses de 80mg à 120mg, contre placebo et paroxetine (56).
Les critères d’exclusion étaient les mêmes que précédemment, avec exclusion supplémentaire des patients prenant une benzodiazépine ou un barbiturique.
Les taux d’abandon n’étaient plus que de 10% à 13% sous duloxétine, et autant sous paroxétine. Les résultats étaient fournis avec et sans correction LOCF. La documentation disponible ne précise pas par quelles mesures (contraintes ou incitations) ou pour quelles raisons le taux d’abandon était 3 à 4 fois plus faibles en Europe de l’Est qu’aux USA.
Effets secondaires réévalués
Ce taux de participation jusqu’au bout de l’étude est d’autant plus remarquable que l’incidence des effets secondaires incluant les défections est, cette fois-ci, mesuré : 46,3% pour l’étude HMAYa, 34,9% pour l’étude HMAYb, ce qui n’avait pas été fait précédemment. En effet dans les études initiales, n’étaient comptés que les effets secondaires déclarés par les participants n’ayant pas abandonné, et les abandons étaient très nombreux (35% à 42% respectivement). Toutefois le laboratoire et la FDA ne publieront pas les incidences de chaque effet secondaire dans ces études complémentaires, et les notices officielles qui seront publiées par la suite dans les différents pays (46)(6)(2) se contenteront de reporter les incidences minimisées par la seule prise en compte des effets supportés sans abandon jusqu’au terme de l’étude.
Revenir au début
Résultats d'efficacité réévalués
L’application des corrections LOCF faisait baisser le résultat de la duloxétine de 1 point, et l’améliorait pour le placebo de ¾ de point. Au total l’avantage de la duloxétine par rapport au placebo, en données corrigées, était plus faible (Tableau 2). L’avantage par rapport à la paroxétine était compris entre 0,5 et 1 point. La FDA a également effectué ses propres calculs avec application de corrections de type LOCF sur les résultats initiaux.

Tableau 2 synthèse des abandons et de leur effet sur les résultats. Source des chiffres : FDA 2003, sur la base des résultats transmis par le laboratoire pour l’ensemble des essais de phase III . (56). * non communiqué ** personnes ayant été au terme de l'étude *** en prenant en compte le dernier résultat connu des personnes ayant abandonné ; **** la dose a été diminuée en cours de traitement pour un nombre non communiqué de personnes.
Bien que reconnaissant que la dose avait dû être diminuée en cours de traitement pour un certain nombre - non communiqué - de patients, le laboratoire estima que, si l’étude HMAYa montrait la supériorité de la dose de 120mg, l’étude HMAYb échouait sur ce point. Il conclut en recommandant à demi-mot une dose de 60mg. La FDA considéra, elle, que la localisation des études HMAY dans les pays de l’Europe de l’Est était responsable, pour des raisons "culturelles", des différences de résultats à dose identique entre les études initiales et complémentaires, et conclut qu’une dose de départ de 40mg à 60mg était recommandée.
Les effets secondaires, incluant cette fois-ci ceux ayant causé l’abandon, étaient présents pour 40% des personnes, sans préciser le chiffre spécifique sous traitement ou sous placebo. Le chiffre spécifique des effets secondaires pour les patients sous duloxetine était donc très probablement supérieur à 40%.
Le laboratoire, dans ces études complémentaires, ne trouva cette fois-ci pas de différence pour les patients avec insomnie ou mélancolie sans préciser leur nombre. Le laboratoire ne donna pas les résultats dans les échelles prévues et autres que HAMD-17, à savoir CGI et PGI, bien que cette dernière ait été annoncée comme mesure secondaire dans les plans d’études. Le Tableau 3 ci-dessous, issu de l'analyse des correspondances entre la FDA et le laboratoire (56), montre les résultats exprimés en échelle PGI.
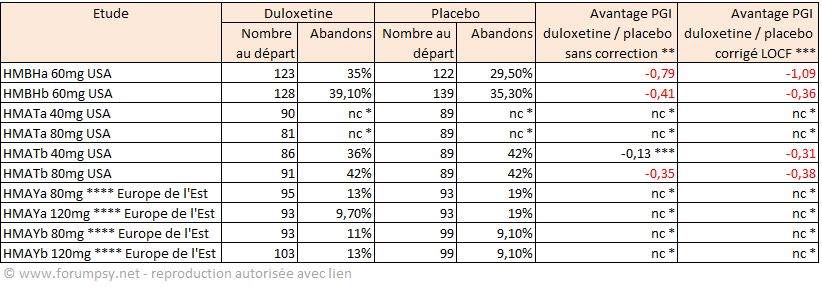
Tableau 3. Synthèse des abandons et de leur effet sur les résultats en terme de ressenti par les patients (échelle PGI). Source des chiffres : FDA 2003, sur la base des résultats transmis par le laboratoire pour l’ensemble des essais de phase III (56) . * non communiqué ** PGI : Patient Global Impression, sur une échelle de 0 à 5, non publié. Un écart négatif signifie que le placebo a un meilleur résultat que la duloxétine ; *** valeur p importante : chiffre peu significatif.
En 2011, Lilly cherche encore à présenter des chiffres favorables
Le laboratoire sponsorise une étude de Ralitza Gueorguieva, PhD, Craig Mallinckrodt, PhD, and Dr John H. Krystal, MD (62), visant à étudier deux types de trajectoire : les "répondeurs" vs les "non répondeurs". Mais, si les chercheurs parviennent à séparer ces catégories parmi ceux prenant l'antidépresseur, ils n'y parviennent pas pour le placebo. Ils en concluent que l'évolution des "répondeurs" au produit est meilleure que celle du placebo, et constatent que l'évolution des "non répondeurs" au produit est mauvaise en comparaison du placebo.

Evolution à 8 semaines réanalysée en 2011 (62)
En termes simples, cette étude nous enseigne que le placebo permet toujours une amélioration, certes plus lente, mais que l'antidépresseur est, dans 30 % des cas, largement moins bon que le placebo.
Il n'y a toujours aucune analyse sur le long terme de l'évolution des personnes entre placebo et antidépresseur : elle semble bien être meilleure pour les personnes sous placebo, ceomme nous le décrivons dans un autre article (63).
Conclusion de l'efficacité sur le trouble dépressif majeur
- que des études les plus favorables HMBHa, HMBHb, HMATb et HMAYa,
- non corrigées par les données manquantes, que ce soit en terme d’efficacité pour les trois premières, ni en terme d’incidence réelle d’effets secondaires, pour aucune des études,
- en ne faisant pas état des échelles CGI et surtout PGI, cette dernière représentant le point de vue du patient sans qu'il sache s'il s'agit du produit ou d'un placebo, et montrant une supériorité pour le placebo,
- sans analyser l’effet sur les conduites suicidaires, exclues d’emblée des sélections : ce point ne fera l’objet d’une évaluation à grande échelle qu’en 2010, à l’initiative du Royaume-Uni suite au constat d’un grand nombre de tentatives de suicide sous duloxétine (53).
Finalement, la FDA approuva le Cymbalta en 2004 dans l’indication de traitement du trouble dépressif majeur, et fut suivie aussitôt par le monde entier. Jusqu’à ce jour les notices destinées aux médecins aux USA, au Canada et en Europe (46)(6)(2), ne font généralement état :
Revenir au début
A nos lecteurs
Nous sommes réceptifs à toute remarque sur le présent article dans le but d'améliorer nos connaissances, et celles des nombreux lecteurs d'un tel sujet. Aussi, n'hésitez pas à "répondre" à ce sujet, ou à nous contacter pour nous signaler toute information utile, remarque, observation, que nous étudierons dans les meilleurs délais.
L'équipe Neptune
V. Notes et références
Revenir au début
